
| BIOLOGIA | Desentrañando los misterios del mal de Alzheimer: una enfermedad relacionada a alteraciones en la conformación de proteínas |
RESUMEN
La enfermedad de Alzheimer es la demencia mas común en el adulto mayor. Un evento clave en esta enfermedad es la formación y depósito de agregados proteicos conocidos como amiloide en el cerebro de las personas afectadas por este mal. En este artículo describo nuestros recientes esfuerzos por entender las bases moleculares de la formación de amiloide y la utilización de este conocimiento básico para el diseño de una nueva estrategia terapéutica. Nuestros resultados sugieren que el paso clave en el depósito del amiloide es el cambio en la conformación de un péptido de 4 aminoácidos que, al adoptar una estructura patológica, se convierte en neurotóxico e induce la muerte neuronal. Para evitar y revertir esta alteración conformacional, hemos diseñado unos péptidos pequeños, que llamamos péptidos anti-hoja-ß. Estos son capaces de inhibir la formación de amiloide y de disolver fibras pre-formadas en un tubo de ensayo y en el cerebro de animales de experimentación. También son capaces de prevenir la muerte celular inducida por el amiloide en ensayos realizados en neuronas humanas en cultivo. Nuestros resultados abren un promisorio camino para el desarrollo de un tratamiento que podría ayudar a detener la degeneración cerebral observada en los enfermos afectados por Alzheimer.
El incremento de las expectativas de vida ocurrido en este siglo, debido principalmente a la curación de enfermedades infecciosas, ha aumentado la proporción de individuos de la población que presentan enfermedades de ocurrencia en etapas tardías de la vida. Por otra parte, ha quedado claro en las décadas pasadas que la base patológica de la demencia senil común no corresponde a una arterioesclerosis cerebral, sino más bien a una progresiva degeneración neuronal, similar a la descrita inicialmente por Alois Alzheimer en 1907. Surge entonces la enfermedad de Alzheimer (EA), no sólo como la demencia progresiva más frecuente de la tercera edad, sino que se convierte en uno de los principales problemas de salud pública en el mundo.
Se estima que aproximadamente un 10% de los individuos de 65 años padecen de algún grado de enfermedad de Alzheimer. Esta cifra incrementa paulatinamente con la edad, alcanzando un valor cercano al 50% para la población mundial mayor de 85 años. El mal de Alzheimer se presenta preferentemente (90%) en la forma conocida como esporádica, vale decir los pacientes no tienen antecedentes familiares de la enfermedad y al parecer no hay una modificación del material genético que sea responsable de la manifestación de ella. Por su parte, la forma hereditaria se presenta aproximadamente en un 10% de los casos, se manifiesta en edades mas tempranas que la EA esporádica y los síntomas clínicos son mas severos.
Cuadro clínico y diagnóstico de la EA
El síntoma más típico de la enfermedad de Alzheimer es la pérdida progresiva de la memoria de corto alcance, vale decir las personas olvidan cosas que han vivido recientemente; la personalidad y el carácter tienden a modificarse, los pacientes suelen ponerse muy irritables, presentando una gran agitación, con ganas de discutir y cuestionar todo, descuidando su apariencia externa. A medida que la enfermedad avanza, los pacientes presentan problemas a nivel de la memoria de larga duración y el pensamiento abstracto. En las etapas más avanzadas de la enfermedad los pacientes se vuelven confusos y pierden la orientación en el tiempo, en la fecha en que se encuentran, en ubicar el lugar donde viven, en el reconocimiento de familiares, etc. La persona deja de conversar, presenta cambios frecuentes en su estado anímico, sufre de incontinencia y en casos extremos es incapaz de cuidarse por si mismo, terminando finalmente en la postración extrema.
El diagnóstico de la EA se realiza habitualmente evaluando el cuadro clínico y descartando otras causas de demencia. De esta forma se tiene un 80 a 90% de probabilidad de efectuar un diagnóstico adecuado, el cual sólo se confirma mediante el estudio neuropatológico del cerebro una vez que el paciente ha fallecido. Un método certero de diagnóstico en vida podría ser una biopsia cerebral, procedimiento que, en general, no se realiza por los riesgos que involucra. En los últimos años se ha intentado ayudar al diagnóstico mediante el uso de diversas técnicas que permiten obtener imágenes del cerebro y también a través de la identificación de marcadores bioquímicos (es decir sustancias cuyos niveles se encuentren alterados en las personas enfermas) en líquidos biológicos como el plasma o el líquido cefalorraquídeo. Sin embargo, hasta ahora estos métodos sólo ayudan en parte a descartar otras causas de demencia y el diagnóstico se basa fundamentalmente en la evaluación clínica.
Las manifestaciones clínicas son el resultado de una serie de cambios en el cerebro de las personas que sufren de EA. Es característico en el tejido cerebral de los pacientes la degeneración de neuronas localizadas en regiones del cerebro que guardan mayor relación con la memoria. La degeneración neuronal incluye la muerte selectiva de algunas células nerviosas, la pérdida de sinapsis y la disminución en los niveles de ciertos neurotransmisores.
Las otras alteraciones neuropatológicas típicas de la EA, son la presencia de dos tipos de agregados proteicos conocidos como ovillos neurofibrilares y placas seniles. Los ovillos neurofibrilares están compuestos por haces de filamentos pareados de disposición helicoidal, formados principalmente por una proteína del citoesqueleto, conocida como Tau, que se encuentra enriquecida en grupos fosfato. Por su parte, las placas seniles están compuestas en su periferia por prolongaciones neuronales (dendritas y axones) en degeneración y en la parte central por fibrillas de amiloide. El principal componente de las fibrillas de amiloide es un filamento de 6 a 10 nm de longitud, compuesto de subunidades idénticas de conformación ß-plegada, dispuestas en un ordenamiento anti-paralelo.
La formación de amiloide posiblemente desencadena la EA.
En 1984, se pudo solubilizar por primera vez fibrillas de amiloide obtenidas del cerebro de un paciente, pudiéndose separar el amiloide, el cual se identificó como un pequeño péptido hidrofobico de 40 aminoácidos, que fue llamado péptido beta amiloide o ßA. Por medio de técnicas de ingeniería genética, fue posible luego aislar el trozo de material genético que contenía la información para el péptido ßA, pudiéndose determinar que este péptido estaba codificado en el brazo largo del cromosoma 21 formando parte de una proteína más grande, conocida como la proteína precursora del amiloide (PPA).
Existen varias evidencias que sugieren fuertemente que el depósito del péptido ßA es el evento central que desencadena el proceso de la EA. Entre estas evidencias, se puede señalar el hecho de que algunos de los individuos que desarrollan EA de tipo hereditaria, presentan mutaciones en el gen que codifica para la PPA, localizado en el cromosoma 21. Además, se ha encontrado que las personas que presentan el Síndrome de Down o mongolismo, caracterizado por la existencia de 3 copias del cromosoma 21, presentan EA en prácticamente un 100% de los casos y a una edad mucho menor que las personas normales. Esto sugiere que un mayor número de copias del gen de la PPA sería responsable de la temprana aparición de la enfermedad. Por otra parte, estudios en personas con altas posibilidades de presentar la EA, ya sea por tener antecedentes familiares o padecer del Síndrome de Down, han determinado que la primera señal de la enfermedad es la aparición de depósitos del péptido ßA en la corteza cerebral. Finalmente, se ha descrito la creación de modelos animales de la EA, que se han producido sobreexpresando el gen humano de la PPA alterado con mutaciones asociadas a la EA hereditaria. Estos animales desarrollan típicas placas amiloides en el cerebro, las cuales inducen daño neuronal y alteraciones conductuales.
Hace unos años se descubrió que ßA, además de encontrarse en forma de agregados fibrilares en los depósitos de amiloide, está también normalmente circulando en una forma soluble en el plasma y líquido cefalorraquídeo de individuos normales y afectados por EA. Considerando que ßA es un producto normal del metabolismo celular, producido en muchos tipos celulares distintos, una de las preguntas fundamentales en la biología básica de la EA es: Cuál es el mecanismo, y qué factores determinan que un péptido, que normalmente se encuentra en forma soluble, llegue a adoptar una forma fibrilar que resulta ser tóxica para las neuronas?
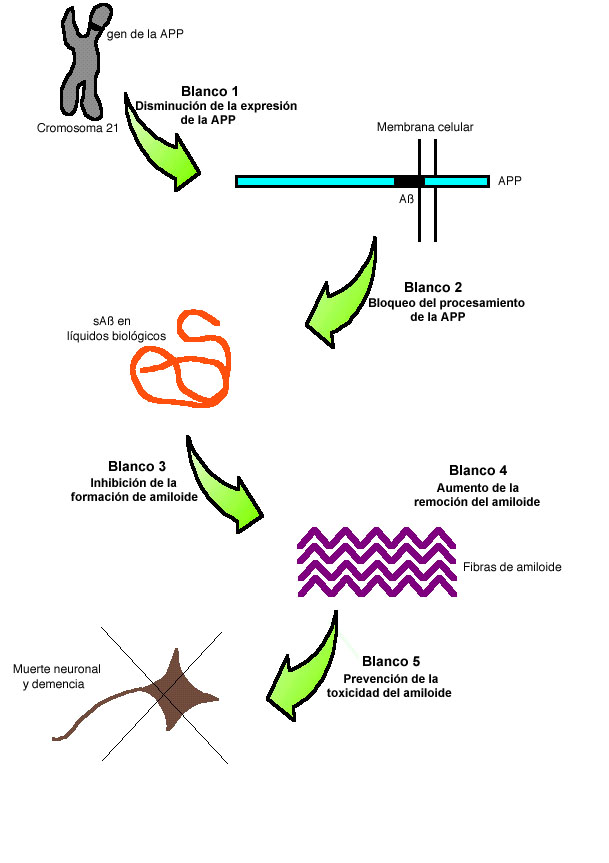
Figura 1
Considerando la importancia del amiloide en la EA, muchos grupos se han dedicado al diseño de estrategias para inhibir la formación de depósitos amiloidogénicos. Existen varios pasos en los que se podría intervenir para bloquear el efecto negativo del amiloide sobre el cerebro (Figura 1): (1) Inhibir la expresión del gen que codifica para la PPA; (2) Disminuir el procesamiento de la PPA que da origen a ßA; (3) Bloquear la formación y depósito de amiloide; (4) Ayudar a la eliminación del amiloide del cerebro y (5) Prevenir el efecto tóxico del amiloide. Nuestro grupo se ha enfocado en la investigación de las bases biofísicas y moleculares de la formación de amiloide con el objetivo de acumular conocimiento suficiente para el diseño de estrategias para prevenir la formación de estos depósitos.
Péptidos sintéticos que contienen la secuencia de ßA han sido muy usados para estudiar la formación de amiloide in vitro, ya que al ser incubados en condiciones apropiadas forman agregados semejantes a fibras amiloides, exhibiendo las características morfológicas y estructurales típicas de ellas. Una estrategia que ha dado muchos resultados para la investigación de la región de ßA que es más importante en la formación de amiloide, ha sido estudiar el efecto de la introducción de cambios en algunos aminoácidos específicos en la secuencia de ßA. Utilizando esta técnica se ha descrito que el reemplazo de aminoácidos hidrofóbicos por otros más hidrofilicos en la porción central de ßA (aminoácidos 17-21), disminuye la tendencia del péptido resultante de dar origen a amiloide, lo cual sugiere que la agregación de ßA es, al menos parcialmente, determinada por interacciones de tipo hidrofóbicas, es decir por la tendencia del péptido de estar lejos del contacto con el agua. La formación de amiloide es también dependiente de las condiciones ambientales. Así por ejemplo, se ha demostrado que el pH (acidez) de la solución determina la velocidad de formación de amiloide. Es importante destacar que en estos experimentos se observó una relación directa entre la modificación de la conformación de ßA en solución y su habilidad para formar amiloide.
La formación de amiloide es determinada por la conformación de ßA.
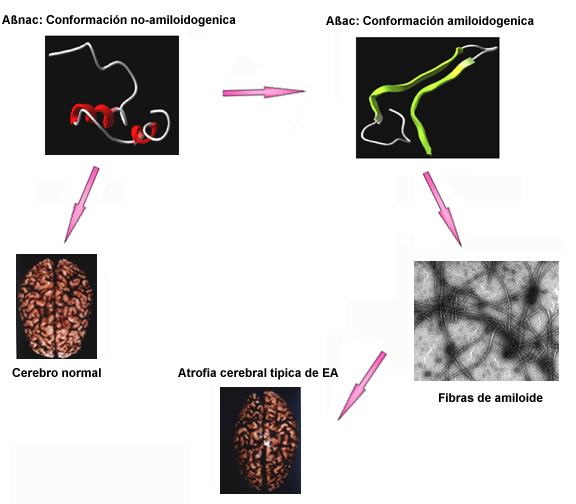
Figura 2
El péptido ßA consiste de al menos dos secciones con diferentes propiedades: un fragmento hidrofóbico que se estructura ordenadamente como lo que se conoce como hoja- y un segmento cuya composición permite que, ya sea se mantenga en forma desordenada, o se estructure como hoja-ß. Nuestra hipótesis de trabajo era que la estructura adoptada por este último fragmento de ßA determinaba las propiedades de agregación del péptido. Para evaluar esta hipótesis analizamos en forma comparativa el efecto de sustituciones puntuales de aminoácidos en esta región sobre la estructura del péptido y su capacidad de formación de amiloide. Con el objetivo de producir un cambio relativo en la tendencia de ßA para ordenarse o desordenarse, se diseñó en forma teórica mutaciones que no produjeran cambios significativos en las propiedades iónicas o hidrofóbicas de ßA, pero que indujeran un cambio detectable de la tendencia de ßA para adoptar la conformación hoja-ß. Un péptido sintético homólogo a la secuencia 1-40 de ßA que contienen una única sustitución (del aminoácido `valina' en posición 18 por `alanina'), mostró un descenso importante en su tendencia a adoptar estructura hoja-ß en comparación al péptido sin modificaciones. Este cambio en la estructura se correlaciona con una disminución notable en la habilidad del péptido modificado de dar origen a fibras de amiloide. Otra substitución aminoacídica que se estudió fue el cambio del aminoácido `ácido glutámico' en posición 22 por `glutamina'. Esta mutación está presente en los individuos que padecen una enfermedad relacionada al mal de Alzheimer conocida como hemorragia cerebral con amiloidosis del tipo Dutch. Un péptido homólogo a ßA que contiene esta substitución forma mucho mayor cantidad, y en forma más rápida, de fibrillas de amiloide que el péptido normal (sin modificaciones), y presenta un mayor porcentaje de estructura hoja-ß. Estos resultados sugieren que la transición conformacional entre la estructura desordenada y la ordenada como hoja-ß modula la formación de amiloide, dando origen a dos especies conformacionales en solución: una capaz y la otra incapaz de formar fibras amiloides. Esta hipótesis predice que ßA se encontraría normalmente como una mezcla de ambas poblaciones. Esta idea se comprobó mediante la separación de ambas especies aprovechándose de sus diferencias en la habilidad para agregarse. Se vio que ambas poblaciones presentan idéntica secuencia de aminoácidos, pero mientras una de ellas tenía una estructura desordenada la otra se ordenaba en forma de hoja-ß. Estos resultados nos permiten proponer que la secuencia de aminoácidos de ßA posibilita la adopción de dos estados conformacionales alternativos y que la forma soluble de ßA que normalmente se encuentra circulando en plasma y líquido cefalorraquídeo humano corresponde a la del péptido con estructura desordenada, que podría de esta manera ser considerado como la forma normal (no tóxica) de ßA. Por otro lado, el estado ordenado en hoja-ß sería la forma patológica de ßA que da origen a los depósitos tóxicos de amiloide en el cerebro (Figura 2). En este escenario es posible especular que la EA podría pertenecer a un grupo recientemente reconocido de desórdenes en los cuales el evento central es la alteración en la conformación de proteínas. Abundante evidencia acumulada en los últimos años sugiere que en otra enfermedad relacionada con amiloidosis (la enfermedad "de las vacas locas" o encefalopatía espongioforme bovina), el agente infeccioso sería solamente una proteína conocida como "prión". Esta también se caracteriza por adoptar dos estados conformacionales alternativos, sólo uno de los cuales esta asociado con el desarrollo de la enfermedad. La forma "normal" se caracteriza por presentar principalmente una estructura de "alfa-hélice, mientras que la forma patológica presenta un grado considerablemente mayor de estructura hoja-ß y aparentemente posee la habilidad de convertir la conformación normal en la patológica. De esta manera, es posible que una característica común a todas las enfermedades relacionadas con la formación de amiloide sea que los péptidos o proteínas que se depositan en las placas amiloides poseen la habilidad de adoptar dos estados conformacionales: uno fisiológico incapaz de formar amiloide y el otro patológico capaz de formar fibras y asociado con el desarrollo de la enfermedad.
Péptidos anti-hoja-ß: una posible estrategia terapéutica para el mal de Alzheimer y desórdenes relacionados a la conformación de proteínas.
Como se discutió anteriormente la evidencia existente indica que la formación de amiloide estaría modulada por al menos dos propiedades intrínsecas de la proteína amiloidogénica: un alto grado de hidrofobicidad y la tendencia a adoptar una estructura de hoja-ß. La hidrofobicidad sería importante para provocar la agregación de la proteína, debido a la necesidad de los fragmentos hidrofóbicos a permanecer lejos del agua. Por su parte, la conformación en hoja-ß es lo que daría a los agregados formados la morfología fibrilar y sus propiedades tóxicas. Nuestra idea fue separar estas dos propiedades en un péptido similar en composición e hidrofobicidad a ßA, pero con una tendencia nula a adoptar estructura de hoja-ß, gracias a la inserción de aminoácidos que desfavorecen la formación de esta estructura. A estos péptidos los llamamos péptidos anti-hoja-ß.
Para diseñar los péptidos anti-hoja-ß nos enfocamos en el fragmento de aminoácidos en posiciones 17 a 21 de ßA, debido a que múltiples evidencias sugieren que este fragmento juega un papel fundamental en la interacción entre moléculas de ßA, la conformación del péptido en solución y la formación de amiloide. Con el objetivo de disminuir la tendencia del péptido a formar parte de estructuras en hoja-ß, introdujimos de 1 a 3 aminoácidos ‘prolina’. La ‘prolina’ es el aminoácido que existe con menos frecuencia en regiones de hoja-ß. La sustitución de prolinas por otro aminoácido es un cambio que está frecuentemente asociado con el origen hereditario de enfermedades relacionadas con la formación de amiloide. Siguiendo estos principios diseñamos un conjunto de péptidos anti-hoja-ß y probamos su actividad inhibidora de formación de amiloide en ensayos in vitro, en cultivos celulares y en modelos animales.
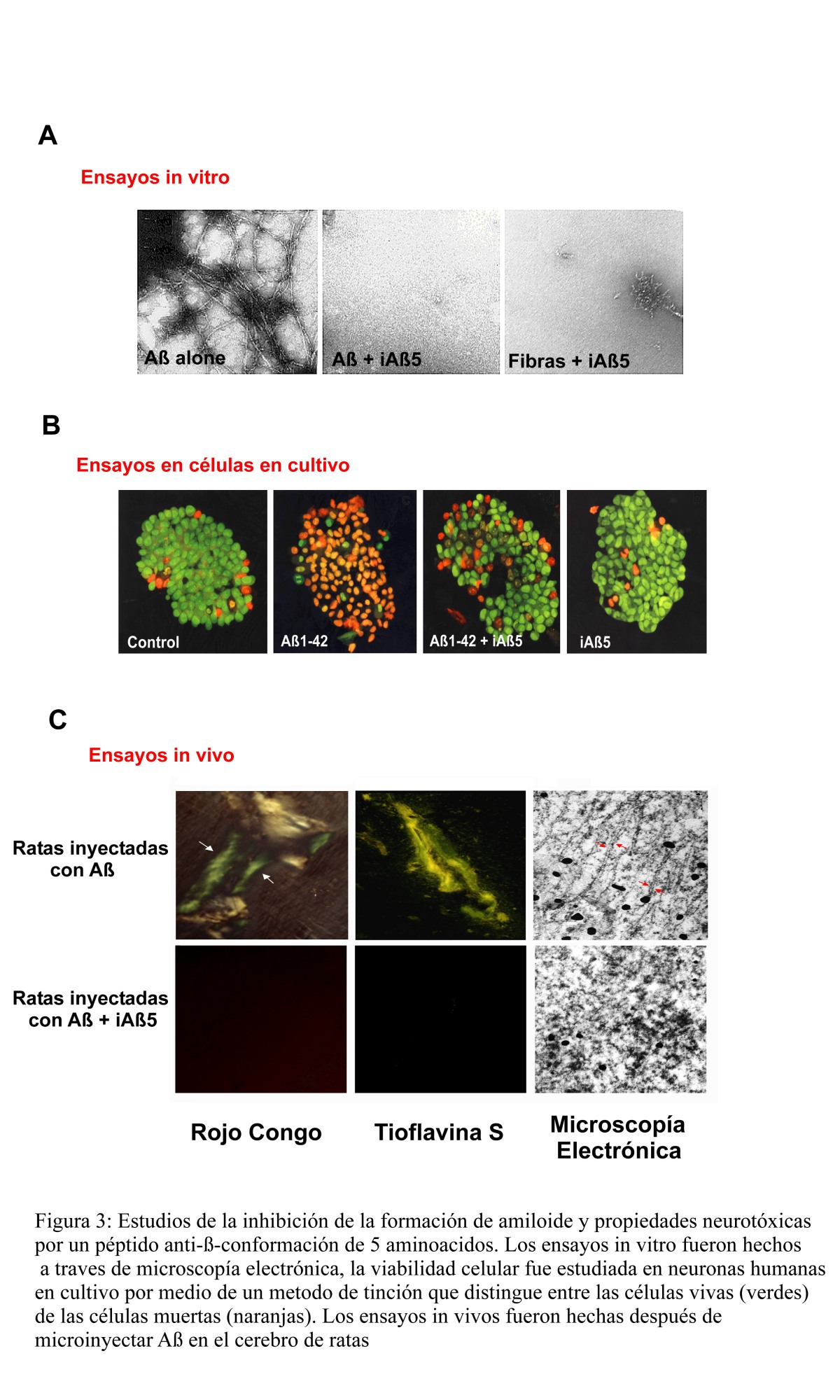
Figura 3
Péptidos anti-hoja-ß de 11, 7 y 5 aminoácidos de largo resultaron ser los mas efectivos en los estudios en tubo de ensayo. En efecto, estos péptidos se unieron con alta afinidad a ßA e inhibieron eficazmente la formación de amiloide in vitro. Además los péptidos inhibidores fueron capaces de desensamblar fibras de amiloide previamente formadas en el tubo de ensayo (Figura 3A). Como se estableció anteriormente, agregados fibrilares de ßA inducen muerte celular en cultivos neuronales humanos. Cuando ßA fue aplicado a cultivos celulares en presencia del péptido anti-hoja-ß de 5 aminoácidos (iAß5), las células prácticamente no fueron afectadas (Figura 3B), sugiriendo que el inhibidor previene la formación del estado neurotóxico de ßA. Finalmente, con el objetivo de probar el efecto de péptidos anti-hoja-ß en el depósito cerebral de amiloide, desarrollamos un modelo animal mediante inyección directa de ßA en el cerebro. Análisis del tejido cerebral de ratas después de 8 días de la inyección, mostró que en el lugar en que el péptido fue inyectado se formaron depósitos de amiloide que exhibían las características morfológicas y bioquímicas típicas del amiloide. En todas las ratas tratadas solamente con ßA se observó la formación de depósitos de amiloide, mientras que en ninguna de aquellas en las cuales se inyectó ßA conjuntamente con el péptido anti-hoja-ß de 5 aminoácidos se observó formación de amiloide (Figura 3C). Esto se puede concluir del análisis histoquimíco del cerebro con colorantes que tiñen específicamente el amiloide y también a través del estudio estructural de los agregados peptídicos mediante microscopía electrónica. Cuando el tejido cerebral fue analizado con agentes que reconocen específicamente ßA (anticuerpos), se observó un alto nivel de ßA en el cerebro de los animales inyectados con ßA, mientras que esta tinción fue aproximadamente 50% menor en las ratas tratadas con el péptido inhibidor. Esto indica que a pesar de que ßA no forma amiloide en el cerebro en presencia del péptido anti-hoja-ß, es todavía capaz de dar origen a agregados amorfos. Es importante destacar que este tipo de agregados amorfos de ßA se encuentran presentes en el cerebro de personas normales de edad avanzada y son por lo tanto considerados no tóxicos. Experimentos en los cuales el péptido inhibidor fue inyectado varios días después de que el amiloide se había depositado en el cerebro de las ratas mostraron que el péptido anti-hoja-ß fue capaz de disolver parcialmente los depósitos, lo cual también resultó en la reversión de alteraciones cerebrales como daño neuronal y activación de microglias.
El conjunto de estos resultados sugiere que péptidos anti-hoja-ß o sus derivados químicos podrían ser útiles para inhibir y disolver la formación de amiloide y de esta manera representan una nueva posibilidad de tratamiento terapéutico para la EA. Recientemente hemos intentado expandir los principios que dieron origen a la generación de los péptidos anti-hoja-ß a otras enfermedades relacionadas a la conformación de proteínas. Para ello elegimos las enfermedades debidas a las alteraciones estructurales de la proteína prión. Diversos resultados in vitro, en células en cultivo y en modelos animales nos permiten concluir que péptidos anti-hoja-ß diseñados de acuerdo a la secuencia de la proteína priónica son capaces de inhibir y revertir los cambios conformacionales de la proteína y el desarrollo de la enfermedad. Estos resultados sumados a los obtenidos en la EA sugieren que los principios para generar péptidos anti-hoja-ß podrían ser aplicados al tratamiento terapéutico de una variedad de enfermedades en las cuales cambios en la conformación de proteínas juegan un papel clave en el proceso patológico.
No hay comentarios:
Publicar un comentario